Aulas Teórico-Práticas de Bioquímica
|
| Bases de Dados |
| Alinhamento de sequências |
| OMIM |
| Visualização de proteínas 2 |
| Árvores Filogenéticas |
Alinhamento de sequências
Neste módulo vamos utilizar uma ferramenta on-line de alinhamento de sequências, o Blast. Esta ferramenta é fornecida pelo NCBI e é de acesso livre. Para este trabalho vamos utilizar a sequência do gene PIPG que obtivemos no módulo de Bases de Dados. Mas pode utilizar qualquer outra sequência que esteja a investigar.
Módulo
1. Abrir o site da NCBI, http://www.ncbi.nlm.nih.gov.
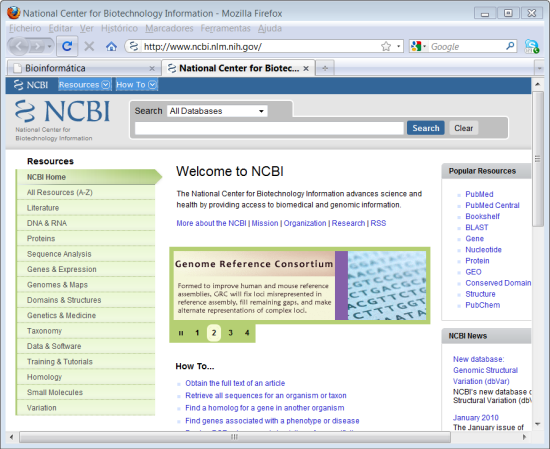
Neste site pode realizar exactamente a mesma pesquiza do site "Entrez" que vimos anteriormente. Repare que no topo temos escolhido a opção "All Databases".
2. Clicar em "BLAST" na coluna do lado direito.
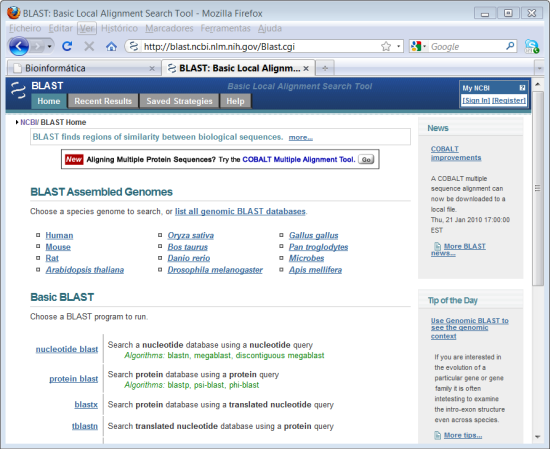
O BLAST ("Basic Local Alignment Search Tool") é na verdade um conjunto de programas para alinhamento tanto de nucleótidos (nucleotide blast) como de proteínas (protein blast).
3. Clicar em "Nucleotide BLAST (blastn).
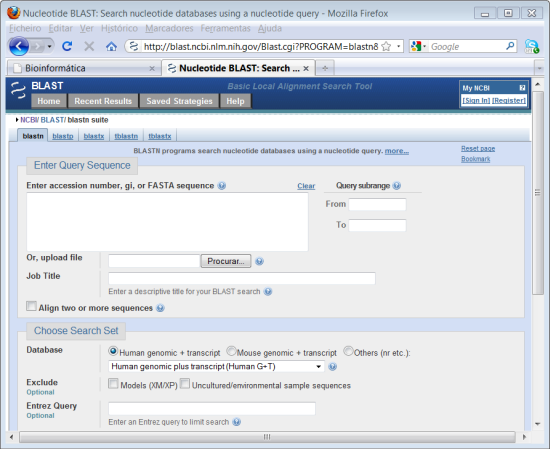
No formulário que aparece no ecrã vai introduzir a sequência obtida no módulo anterior. Nesta página pode definir vários parâmetros para a procura que pretende executar.
4. Abrir o ficheiro de texto que guardou no módulo anterior, faça "copy"da sequência. No campo onde diz "Enter Query Sequence" faça "paste" da sequência.
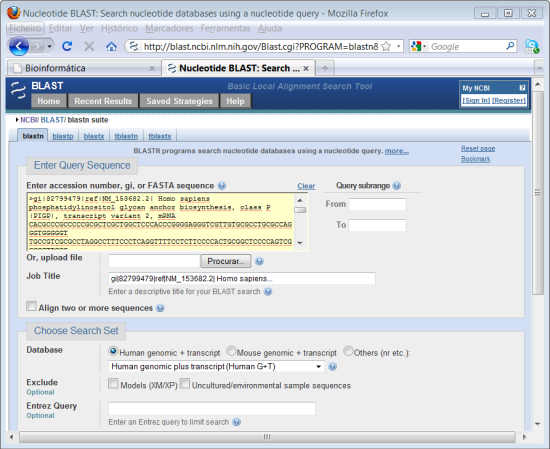
A sequência que colocamos apresenta o cabeçalho característico do formato FASTA em que guardamos a sequência do gene obtido no módulo anterior. Neste caso o programa reconhece o formato da sequência e ignora o cabeçalho não sendo necessário removê-lo. Em alternatica pode simplesmente fazer o upload do ficheiro directamente para o site.
5. Não alterar nenhum parâmetro e primir o botão "BLAST".
O pedido de alinhamento é colocado em lista de espera no servidor BLAST da NCBI. A pesquisa pode demorar algum tempo mas tipicamente demora cerca de 10 segundos. À pesquisa é dada um código "ID" indicado no campo "Request ID".
6. Após alguns segundos aparece a representação esquemática dos resultados significativos. A sequência de nucleótidos do gene que utilizamos para este módulo tem 2457 nucleótidos que estão representados na primeira barra vermelha com a indicação do número de nucleótidos. As barras apresentadas abaixo representam esquematicamente as sequências de nucleótidos que o programa pesquisou que apresentam um grau de homologia mais significativo (da mais significativa para a menos significativa).
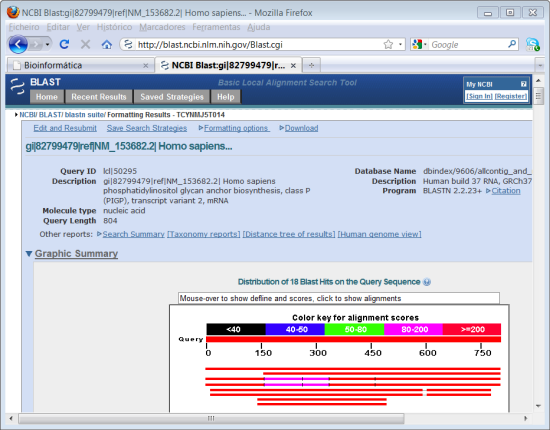
7. Logo abaixo da representação esquemática das sequências com homologia significativa, temos a lista de todos os resultados com a identificação da sequência ("Sequence identifier") e os valores de "Score", de "E", e a percentagem de identidade máxima ("Max Ident") para cada sequência.
O "Score" fornece informação sobre o grau de homologia entre a sequência em questão e a sequência que introduzimos. Quanto maior o valor, melhor será o grau de homologia.
O valor "E" ("Expected") é um indicador do grau de significância do resultado da pesquisa. Quanto mais pequeno o valor de "E" mais significativo é o resultado. O valor de "E" a partir do qual o resultado é significativo varia com o tipo de pesquisa efectuada e os respectivos parâmetros utilizados. No entanto, para uma pesquisa inicial, o valor 0,01 é um bom ponto de partida para aceitar ou rejeitar um resultado como sendo significativo.
Analise os resultados. Quais os resultados mais significativos?
8. Continuar a fazer scroll da página.
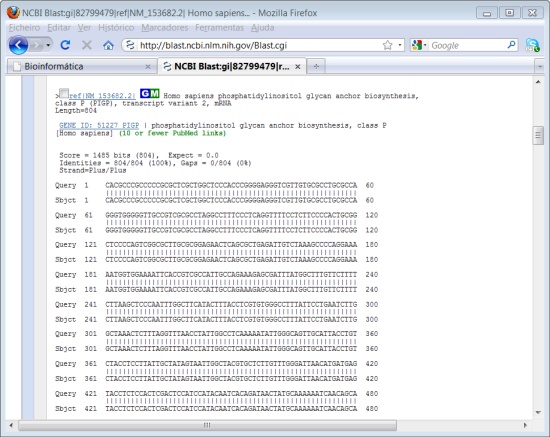
Mais abaixo na página temos o resultado completo do alinhamento para cada um dos resultados. Temos a indicação do "Score". Como o alinhamento é de 100 %, o "E" tem o valor mínimo que é 0 indicando um grau de significância máximo. Como a nossa sequência foi retirada da base de nucleótidos da NCBI o mais provável é que, quando fazemos um BLAST, o primeiro resultado seja a própria sequência.
9. Voltar atrás para a página em que fizeram "copy-paste" da sequência e altere a opção "Program Selection" escolhendo a opção "Somewhat similar sequence (blastn)" em vez da opção "Highly similar sequences (megablast)". Prima novamente em BLAST.
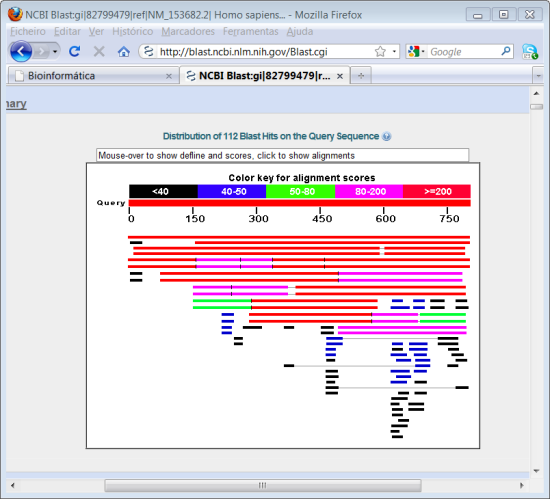
Analise os resultados. Qual é a diferença entre esta pesquisa e a que realizou antes? Compare o número de resultados.